Introduction
The Byos™ disulfide bond analysis workflow comprises identification and quantification of disulfide bonded peptides non-discriminately (expected and unexpected/shuffled). This application note introduces the use of the Byos Disulfide Bond Analysis (S-S) workflow (legacy Byonic™ and Byologic®) for LC/MSMS data, followed by validation, quantification, sample-to-sample comparison, and reporting. Note that accompanying data for a reduced sample is not needed; having only a non-reduced digest is sufficient for analysis.
Summary
- Executing the Byos [S-S] Disulfide workflow
- Configuring the workflow
- Defining Expected Disulfide links (expected)
- Optimizing MS/MS search parameters
- Connecting Sub-Units proteins (inter-chain) or Intra-chain
- Inspecting results and producing a report
Executing the Byos Disulfide (S-S) Workflow
- Click S-S to launch the workflow
- Drag & Drop raw data files
- Drag & Drop a FASTA file
- Select the heavy and light chains from the dropdown menus. The list of expected disulfide links will populate.
Note: The Expected disulfide links table is editable. Any S-S bonded peptide within this table will be categorized as “Expected” in the Byologic peptides table, others will be still be identified but marked as “Shuffled”. - Create a project
Configuring the Workflow
MSMS Identification using Byonic Node
Byonic can readily identify disulfide bonded peptides from a non-reduced digest. Please refer to the below settings for a typical disulfide bond search set up and descriptions
Sample digestion options:
Cleavage site(s): “RK” for trypsin;
“RK;DE” for trypsin+Asp-N cocktail
Cleavage side: “C-terminal” for trypsin;
“C-terminal;N-terminal” for trypsin+Asp-N cocktail
Digestion specificity: Fully specific is recommended. Semi-specific searches will search for semi-specific base peptides, but the partner peptide will always be fully specific. Non-specific is not supported by disulfide bond analysis. If a non-specific enzyme is used for digestion, enter the most common cleavage sites explicitly (eg. FLWY) and set this option to semi-specific.
Missed cleavages: “3” or more. Disulfide bonds typically hinder proteolytic digestion by common enzymes, hence we recommend 3+ missed cleavages.
Modifications:
A recommended list of modifications are:
Oxidation / +15.994915 @ M, W | rare1
Deamidated / +0.984016 @ N | rare1
Gln->pyro-Glu / -17.026549 @ NTerm Q | common1
Disulfide bonds are “rare” type modifications. It is possible to search for modifications on disulfide bonded peptides, though care must be taken in common/rare designation and total max values. If the total rare max value is 1, as in the example above, and Oxidation, and Deamidation is set as rare as well, Byonic will not search for Oxidation or Deamidation on modified peptides. However, because Q->pyroQ is set as common1, Byonic will search pyroQ modification on disulfide bonded peptides.
Note that modifications are searched only on base peptides, and not considered for partner peptides, although Byonic considers both peptides in each role, hence it will identify disulfide bonded peptides with one modified peptide.
If the sample is alkylated (although not reduced), you may include the alkyalting agent as variable modification, eg. common2.
S-S, Xlink
“Enable Disulfide” option is already checked in the S-S workflow. If trisulfide bonds are also of interest, check the “Enable Trisulfide” option.
Enter protein position number in the FASTA in “For FASTA proteins” field. Separate numbers using comma, and separate groups by semi-colon.
Examples: “1” searches for disulfide bonds in the 1st protein in the fasta file. This includes any peptide containing cysteine to any other peptide containing a cysteine, including itself, multiple S-S bonds between two peptides, and intra-peptide disulfide bonds. “1,2” searches for inter chain S-S bonds betweeen protein 1 and 2, and intra-chain S-S bonds in proteins 1 and 2. TIP: This is the common setting for monoclonal antibodies. “1,2;3” searches for crosslinks on the 1st, 2nd, and 3rd proteins. Proteins 1 and 2 can crosslink to each other, but not to protein 3.
Inspection and quantification
Once identification is complete, a project will be created to validate identified peptides, categorize S-S bonds as Expected or Shuffled, compare across samples, and produce a comprehensive report. Below is a step-by-step list of best practices for inspecting the results.
- Use filters in the project window to rapidly identify and eliminate false positives
-
- In the peptides table, if the disulfide bond is present along with another modification, and only with another modification, mark it as false-positive (Ctrl+F)
- Inspect the MS1 isotopic profile and find the blue dot. Is the blue dot on the monoisotopic peak? If not, most likely there is an off-by-one error in the precursor assignment. Mark as false-positive.
- Inspect MS2. Fragments ions from both the base and the partner peptide should be annotated. Partner peptide ions will be indicated with a “(2)” next to the ion label, as below.
Byonic scores both the base and partner peptide. Base peptide score can be found under “Score” column, and partner peptide in “X-link Score” column. A low X-link score may or may not indicate a false positive ID. If the Cysteine is near the C-terminus, y-ions may be missing from the spectra, and hence create a low, possibly negative score. Byonic scores reflect the number, intensity, and m/z errors of fragment peaks; a low X-link score doesn’t necessarily indicate a false positive, because a correct base peptide with a good score, along with a precursor mass matching a disulfide bonded pair is often sufficient to make an identification in one-protein samples.
- [OPTIONAL] If an S-S link has been identified in some of the samples, but not in some others, you can still generate an XIC and thus be able to quantify the peptide in the unidentified sample. Click on “Edit -> In-silico Peptides -> Add Missing via Existing Peptides…”
Reporting
-
-
- Generate a report by clicking the report icon in the toolbar, or go to “File -> Export -> Report”
- The S-S report template is automatically loaded for Byos-created projects. If not, go to File à Presets -> Report Presets -> Blgc_Std_S-S_Profile.rptc
- This report configuration includes:
- A summary tab with protein coverage
- A detailed pivot table with relative abundances of XIC areas of all disulfide bonded peptides.

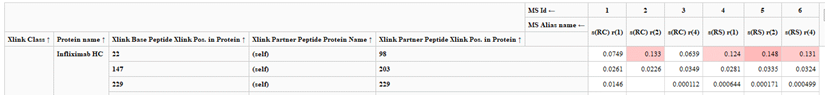
- TIP: If you would like to sum XIC areas of all possible Cys22-Cys98 linked peptides, simply remove the Mod. Names column:
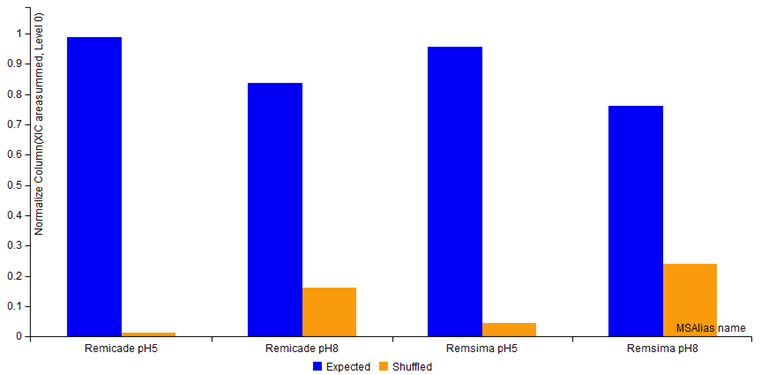
- A bar chart comparison of total expected vs. shuffled S-S bond XICs.
- A bar chart comparison of total disulfides vs. trisulfides.
- A table which groups peptides, disulfide bonded and others, based on the location of the Cysteine residue in the protein sequence.
-
- If you would like to include spectrum and XIC images, click “Tabs -> Add Plots”. This step can take a few minutes depending on the number of peptides that you have.
- Click “Export->Export to PDF…” to generate a single pdf file with all the tabs.
- For more information on how to customize this report, please see our related videos or contact us.