With thanks to:
- M Ford, MS Bioworks, LLC, Ann Arbor, MI, U.S.A.
- S Saveliev, Promega Corporation, Madison, WI, U.S.A.
- K Pisupati, R Ackermann, A Schwendeman, Biointerfaces Institute, University of Michigan, MI, U.S.A.
- Protein Metrics Customer Success Team, Cupertino, CA, U.S.A
Summary
- This Technical Brief illustrates the application of the Byos Disulfide workflow (S-S) to a stability study of Infliximab, marketed as the innovator biotherapeutic Remicade and a biosimilar Remsima.
- Within Byos, automated labeling of identifications using both the Disulfide Validators and Xlink labels improves the efficiency of data review.
- For organizations that share information across teams or different laboratories, results from the disulfide analysis can be uploaded to Byosphere for ease of view in any browser at any location.
- Organizations that adopt these workflows benefit from consistency across different mass spectrometry platforms and laboratories worldwide.
Introduction
Unpaired cysteines or variant disulfide bonding can adversely affect monoclonal antibodies (mAbs), or other biotherapeutic proteins, and hence there is significant interest in mapping and quantifying disulfide variants down to low concentrations. Knowledge of the disulfide bond patterns also provides invaluable information for faster process development. Furthermore, monitoring disulfide bonds is critical for confirming product and process consistency.
Data from modern mass spectrometers can determine the disulfide bonds but are complex and time-consuming to interpret.
Key challenges faced have included:
- The need for highly expert users to interpret the analysis
- The astronomical number of potential ‘scrambled’ bonds
- Need to quantify both disulfide and trisulfide variants down to low concentrations
- The huge time burden for manually processing all possibilities, even for expert users
With the challenges faced, comprehensive, manual interpretation of all unpaired cysteines and disulfide bond variants is impractical.
Here we present an automated method for rapidly characterizing and quantifying disulfide bonds, including their shuffling. Application is made to innovator (Remicade) and biosimilar (Remsima) versions of infliximab. Included is an example of how Byos can automatically flag results based on a decision tree specifically designed for unpaired cysteine and disulfide bond analysis. This automated labeling system can be used to vastly improve efficiency of data review.
Experimental Conditions
Two different infliximab monoclonal antibodies were sourced for this study: the originator drug, Remicade (Janssen Biotech Inc.) and the biosimilar Remsima (Celltrion Inc.).
Two different conditions for each biologic drug were assessed:
- pH 5: At this pH, the free cysteines should be protonated and less likely to induce shuffling of the disulfide linkages
- pH 8: At this pH, free disulfides are much more likely to be deprotonated and disulfide shuffling is more likely to occur.
Each of the samples of infliximab monoclonal antibody were digested under non-reducing conditions and analyzed by LC-MS/MS on a Thermo Q-Exactive high resolution mass spectrometer.
Typical parameters for set up of the disulfide workflow are shown in Figure 1 and the setup is outlined in more detail in the Disulfide Bond Analysis Application Note1.
The search engine within Byos searches for, and displays, disulfide-bonded peptides and was applied to data on the innovator product as well as the biosimilar product. Disulfide-bonded and trisulfide-bonded peptides with abundances varying over more than three orders of magnitude were assigned. The automated report generated includes plots that visualize the data across samples, providing relative quantification of the species identified, and pinpointing exactly which locations differed between the two products.
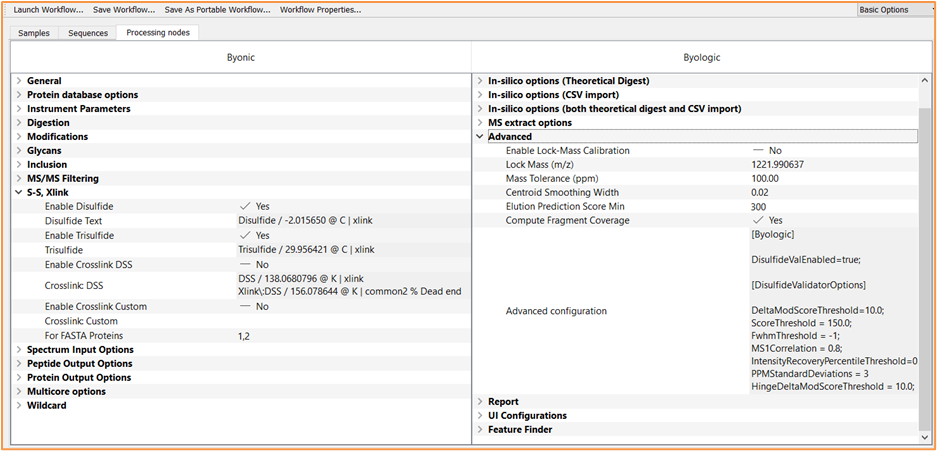
Figure 1: Byos project creation window from the automated workflow for disulfides and cross-linked peptides within Byos. Note the inclusion of “DisulfideValEnabled=true” within the advanced configuration of Byologic. This command is all that is required to turn on the disulfide validators. If the user would like to change any of the default parameters, the [DisulfideValidatorOptions] can be added and modified as required.
Results and Discussion
Results from disulfide analysis
In addition to the output from typical peptide mapping experiments, the disulfide analysis results include additional parameters with the prefix “Xlink” that can be used to review, filter, and report the data. Figure 2 shows three of these columns, along with other output from the resulting project.
The review process can be streamlined by following some key principles that are well understood in disulfide bond analysis. A key point to note is that it is very rare for “Expected” disulfide pairs to be incorrectly identified. The review time for “Expected” should therefore be minimal and the largest part of data review spent on “Shuffled” disulfides. The automatic output from Byos allows for these categories to be filtered in or out to expedite the review process.

Figure 2. Resulting output from the automated Byos workflow for disulfide bond analysis of Remicade and Remsima. The figure shows the peptide table with one species selected (A), the MS/MS spectrum (B), extracted ion chromatogram (C) and MS spectrum (D) of the selected disulfide bonded pair. Visualization of the data in a single view allows for rapid review of the data. The inset shows how the data can be filtered by any column by selecting the filtering icon. By filtering on the “Xlink Class” column, expected disulfides can be filtered out to focus review time on shuffled disulfides.
A previous application note1 outlines some steps for reviewing disulfide data. The recent addition of the disulfide validator labels in the “Comments” column of the peptide table add another tool to improve the efficiency of data curation. The logic used to label each result in the comments column is shown in Figure 3. The logic process produces warning flags rather than act as a ‘black box’, enabling a user to intervene if needed.

Figure 3. Logic used for labeling of disulfide bond variants when the validator algorithm is deployed in Byos. Variants with a ‘critical’ label are highly likely to be false positives. Those with a ‘warning’ label should be manually inspected. Those variants labeled ‘clear’ are highly likely to be true positives and should not require significant review time. Combining these comments with other information from the peptide table, reduces the data curation burden significantly. The default threshold values for RT Diff, the various peptide scores, MS1 Correlation, Mass Accuracy and Recovery are shown above. These threshold values can be changed to analysts’ preference by changing the values in the advanced command.
*Note: The FWHM is used as the minimum retention time difference that categorizes a modification as not co-eluting with its wildtype. A setting of -1 determines the average FWHM of the input files and then automatically calculates the threshold. Alternatively, this can be set manually using any real number greater than 0.
As shown in Figure 4, each peptide identification is automatically labeled with ‘critical, ‘warning’ or clear’ in the ‘Comment’ column of the peptide table (A) to enable rapid data review and to easily filter data for reporting. The highlighted example in Figure 4 shows the label “Warning: Score < Threshold”.

Figure 4: Example results from disulfide analysis with disulfide validators enabled through the Advanced configuration option. The peptide table (A) includes additional information in the “Comment” column to aid data review. The example highlighted shows “Warning: Score < Threshold”. As can be seen from the MS2 data (B) there are some ions indicating that this is a true match, but the data should be verified manually. The chromatogram (C) contains neighboring red dots that shows multiple MS2 scans with the same ID, and the MS1 data (D) has an isotope prior to that of the identified peptide mass (blue dot on 867.419). Checking the data from this peptide identification in other injections helped to support that it was a true positive. The disulfide validators within Byos enables the user to focus on verification of those identifications that should be manually checked.
Along with using the comments column, results can be assessed by their response, score (the base peptide score) and Xlink score (the partner peptide score). Using this information, false positives can be rapidly labeled as such by selecting groups of results and pressing Ctrl + F such, as described in an early application note1.
Automated reporting from disulfide analysis
When the data is processed using the Byos |-S-S-| workflow, an interactive report is automatically generated. The default report configuration for |-S-S-| contains five tabs: The ‘S-S Summary Tab’, which shows an overview of the protein sequence where modified amino acids are highlighted in a different color; the “S-S Compare” tab which lists all identified bonded cysteines and their partner peptides by extracted ion chromatogram intensity; the “Expected versus Shuffled Barchart” tab (shown in Figure 5); the “Di- vs Tri-sulfides Barchart“ tab (shown in Figure 6); and the “S-S Compare by Cys Loc” tab (shown in Figure 7). As the results are curated in the Byos project, the report reflects the results from the project. Reporting tabs can be modified, and additional tabs created to reflect individual user requirements.
The bar chart in Figure 5 clearly displays the similarities and differences between the disulfides detected in each of the samples. As expected, higher levels of disulfide shuffling were apparent in both the Remicade sample and the Remsima sample at pH 8 compared to pH 5.

Figure 5: Results from the ‘Expected vs Shuffled Barchart” tab which is automatically generated in the default Byos report for the |-S-S-| workflow. An “Expected” cross-linkage is a defined linkage in the “Disulfide links (expected)” table during project creation. This barchart shows the sum of the extracted ion chromatogram responses from all expected disulfides/trisulfides (“Expected” shown in orange) and the sum from all shuffled disulfides/trisulfides (“Shuffled” shown in blue). As would be expected, in both the originator drug (Remicade) and the biosimilar (Remsima), a higher response from shuffled disulfides is apparent under the higher pH conditions (pH 8 compared to pH 5).
The barchart in the “Di- vs Tri-sulfides Barchart“ tab shown in Figure 6 automatically calculates the contribution of di- tri- or free thiol peptides. As would be expected from these samples, the majority of the response of cysteine containing peptides comes from disulfide-bonded peptides. There is, however, a small contribution from both trisulfides and free-thiol peptides, which can easily be seen by toggling the disulfide category label (Figure 6A versus Figure 6B).

Figure 6: Results from the ‘Di- vs Tri-sulfides Barchart” tab which is automatically generated in the default Byos report for the |-S-S-| workflow. The barchart shows the summed area of disulfides, trisulfides and free thiols for all cysteine containing peptides as a normalized percentage. The barchart with all categories selected (A), shows that the majority of the response from cysteine-containing peptides is from those involved in disulfide bonding (orange columns). The large response from disulfide bonded peptides makes it difficult to see the response from free thiol and trisulfide peptides. This is easily changed by clicking on the disulfide category label, which then hides that data so the response from other categories can be seen (B).
Disulfide shuffling is a key concern for the stability, efficacy, and safety of biotherapeutics and is expected to be more apparent at higher pH. Figure 7 clearly shows the impact of pH on disulfide shuffling for one example in both Remicade and Remsima. In all samples, the highest response was from the expected disulfide form of the peptide at Cys22. With the higher pH samples, however, there was clear an increase in the shuffled forms that included Cys22. This was also apparent for other cysteine locations across the two biotherapeutics (data not shown).

Figure 7: Excerpt of the results from the ‘S-S Compare by Cys Loc” tab which is automatically generated in the default Byos report for the |-S-S-| workflow. The table is presented as a heatmap whereby each cysteine-containing peptide is normalized based on the total response for all cross-linked peptides at that location. The colors in this heat map have been changed so that the highest contributors to the extracted ion chromatogram response are shown in blue. Peptides of lower abundance are shown in shades of orange. In the example here, peptides containing the cysteine at position 22 and its partner peptides are shown. For ease of viewing, the percentage response values have been expanded. The first peptide (shown in dark blue), which is an expected disulfide pair, contributes the highest response for all identifications that include residue 22. The other contributors to overall response at this location are much lower. The heatmap quickly shows that the samples at pH8 (injection 1 and 3) have a higher contribution from the shuffled disulfide pairs, as shown by the orange shading.
Disulfide analysis within Byosphere
For organizations where the analysis of disulfides is shared across multiple laboratories or teams, Byosphere enables enterprise-wide collaboration. Analysts can manage, interrogate, and share files and reports generated from any analytical lab using Protein Metrics software. Results can be uploaded to the server, as shown in Figure 9, and the report viewed in any browser. If required, other users can be granted access to download the project or report for deeper interrogation of the data, as shown in Figure 10.
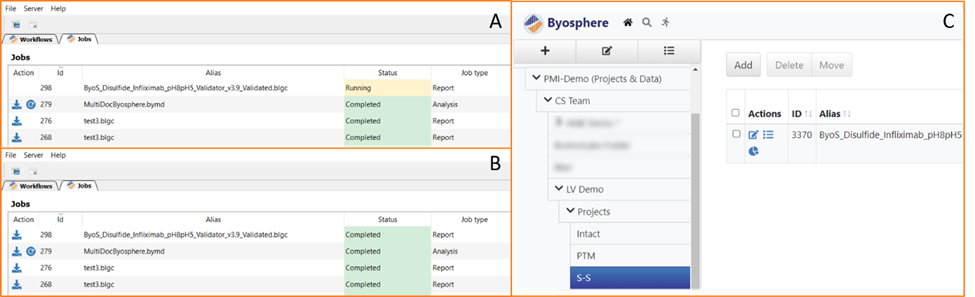
Figure 9: Uploading results to the Byosphere server. When a report or analysis is uploaded to Byosphere, it is shown in the queue. First as ‘Queued’ (not shown) then as ‘Running’ (A) and finally as ‘Completed’ (B). Once complete, the report can be viewed in any browser (C).
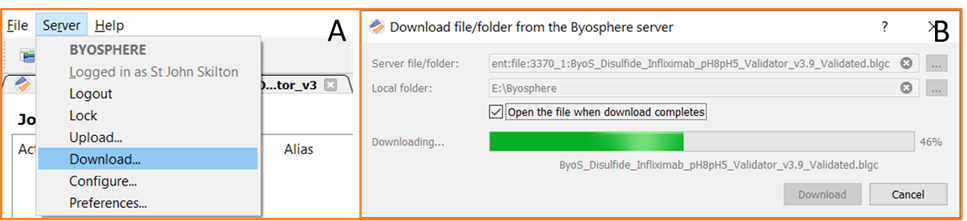
Figure 10: If advanced users require access to change or review the data, they can download the file from the server.
Conclusions
Byos offers a platform and workflow for the efficient identification, inspection, and relative quantification of disulfide bonds pairs (both expected and unexpected). Trisulfide bonds and free cysteine residues can be identified and quantified at the same time.
The detailed, quantitative comparisons for both expected disulfide bonds and shuffled disulfide bonds provides an in-depth understanding of the product and can significantly enhance product development.
Data sharing and collaboration across organizations can be vastly improved by deploying Byosphere, either with processing on-premise or in the cloud.